Benchmarking tokenizers for cross-tissue generalization#
Here, we demonstrate how to use Heimdall for benchmarking the impact of tokenizer choice on cell-type annotation performance in a challenging cross-tissue evaluation setting. For this evaluation, we use a subset of the scTab dataset.
import hydra
import Heimdall
from matplotlib import pyplot as plt
import matplotlib
import seaborn as sns
import scanpy as sc
plt.rcParams['pdf.fonttype'] = 42
plt.rcParams['svg.fonttype'] = 'none'
sc.set_figure_params(figsize=(6, 6), frameon=False)
sns.set_theme(style="white")
Recap: the Heimdall framework#
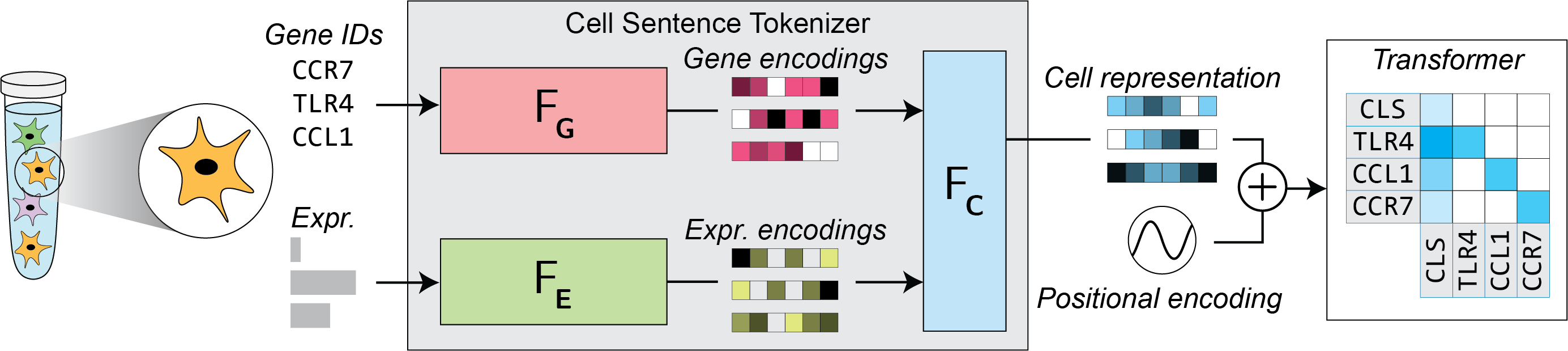
We are interested in implementing various popular tokenizers for single-cell foundation models within a standardized framework, which enables us to isolate the impact of the tokenizer itself on downstream performance.
Experiment setup (via hydra)#
We use hydra to configure each Heimdall run. First, we detail the configuration files that are shared across all tokenizers for this cross-tissue generalization experiment.
Let’s take a closer look at some of the most important configs…
experiments#
Top-level experiment config that specifies other essential configs.
defaults:
- override /dataset: new_sctab # dataset for the relevant task
- override /tasks: new_sctab_split # dataset for the relevant task
- override /model: transformer # the chosen model
- override /scheduler: cosine # contains scheduler
- override /trainer: default # contains optimizer and trainer details
- override /optimizer: AdamW
- override /fg: random
- override /fe: noop
- override /fc: geneformer
seed: 55 # random seed for reproducibility
project_name: new_sctab_split1 # project name for WandB
dataset#
Specifies the path to the dataset, as well as preprocessing arguments.
dataset_name: new_sctab
preprocess_args:
data_path: ${data_path}/sctab/tissue_splits_spencer/scTab_GItract_train.h5ad
top_n_genes: false
normalize: true
log_1p: true
scale_data: false
species: human
task#
Specifies the task to use for training the model, including training metrics, dataset splits, the architecture of the task “head” (used for predicting the task outputs), and the loss function. In this case, the predefined dataset splits (stored in the adata.obs["split3"] column) reserve gastrointestinal tract cells exclusively for training, while brain cells are used exclusively from validation/testing.
type: Heimdall.task.SingleInstanceTask
args:
task_type: multiclass
label_col_name: cell_type
metrics: [Accuracy, MatthewsCorrCoef, ConfusionMatrix]
track_metric: MatthewsCorrCoef
splits:
type: predefined
col: split3
keys_:
train: train
val: val
test: test
early_stopping_patience: 5
early_stopping: true
shuffle: true
batchsize: 32
epochs: 50
dataset_config:
type: Heimdall.datasets.SingleInstanceDataset
head_config:
type: Heimdall.models.LinearCellPredHead
args:
loss_config:
type: Heimdall.losses.FlattenCrossEntropyLoss
cell_rep_config:
type: Heimdall.cell_representations.CellRepresentation
model#
Specifies the model architecture used for the scFM.
type: Heimdall.models.Transformer
name: transformer
args:
d_model: 128
pos_enc: BERT
num_encoder_layers: 2
nhead: 4
hidden_act: gelu
hidden_dropout_prob: 0.1
use_flash_attn: false
pooling: cls_pooling # or "mean_pooling"
Modular reimplementation of the Geneformer tokenizer#
Having configured everything except the tokenizer, we now focus on implementing the tokenizer. For practice, let’s implement the Geneformer tokenizer.
fg - the gene identity encoder (\(F_\textbf{G}\))#
Specifies the Fg implementation for this tokenizer, as well as the torch.nn.Module used for providing the embeddings of the genes. Here, we use the random implementation, which assigns a randomly-initialized embedding vector of dimensionality model.args.d_model to each gene in the cell.
type: Heimdall.fg.IdentityFg
args:
embedding_parameters:
type: Heimdall.embedding.FlexibleTypeEmbedding
args:
num_embeddings: vocab_size
embedding_dim: ${fg.args.d_embedding}
d_embedding: ${model.args.d_model}
frozen: false
fe - the gene expression encoder (\(F_\textbf{E}\))#
Specifies the Fe implementation for this tokenizer, as well as the torch.nn.Module used for providing the embeddings of the genes’ expression levels. Here, we use the noop implementation, which simply outputs a vector of zeros of dimensionality model.args.d_model for each gene in the cell, regardless of the gene’s expression level.
type: Heimdall.fe.IdentityFe
name: Heimdall.fe.IdentityFe
args:
embedding_parameters:
type: Heimdall.embedding.ZeroBroadcast
args:
out_features: ${fe.args.d_embedding}
d_embedding: ${model.args.d_model}
drop_zeros: true
fc - the single-cell representation function (\(F_\textbf{C}\))#
Specifies the
type: Heimdall.fc.Fc
args:
max_input_length: 2048
embedding_parameters:
type: torch.nn.Module # Should throw an error if called
tailor_config:
type: Heimdall.tailor.ReorderTailor
order_config:
type: Heimdall.order.ExpressionOrder
reduce_config:
type: Heimdall.reduce.SumReduce
Putting it all together#
from omegaconf import OmegaConf
with hydra.initialize(version_base=None, config_path="../Heimdall/config"):
config = hydra.compose(
config_name="config",
overrides=[
"+experiments=sctab_split1_all",
"fg=random",
"fe=noop",
"fc=geneformer",
],
)
OmegaConf.resolve(config)
print(OmegaConf.to_yaml(config))
project_name: new_sctab_split1
run_name: Heimdall.fg.IdentityFg_Heimdall.fe.IdentityFe_Heimdall.fc.Fc_Heimdall.models.Transformer_lr0.002_bz32
work_dir: new_sctab_split1_results/Heimdall.fg.IdentityFg_Heimdall.fe.IdentityFe_Heimdall.fc.Fc_new_sctab_lr0.002_bz32_seed55_agTrue
run_wandb: true
float_dtype: float32
seed: 55
data_path: /work/magroup/shared/Heimdall/data
ensembl_dir: /work/magroup/shared/Heimdall/data
cache_preprocessed_dataset_dir: /scratch/heimdall/shared/cache
entity: Heimdall
only_preprocess_data: false
model:
type: Heimdall.models.Transformer
name: transformer
args:
d_model: 128
pos_enc: BERT
num_encoder_layers: 2
nhead: 4
hidden_act: gelu
hidden_dropout_prob: 0.1
use_flash_attn: false
pooling: cls_pooling
dataset:
dataset_name: new_sctab
preprocess_args:
data_path: /work/magroup/shared/Heimdall/data/sctab/tissue_splits_spencer/scTab_GItract_train.h5ad
top_n_genes: false
normalize: true
log_1p: true
scale_data: false
species: human
tasks:
type: Heimdall.task.SingleInstanceTask
args:
task_type: multiclass
label_col_name: cell_type
metrics:
- Accuracy
- MatthewsCorrCoef
- ConfusionMatrix
track_metric: MatthewsCorrCoef
splits:
type: predefined
col: split1
keys_:
train: train
val: val
test: test
early_stopping_patience: 5
early_stopping: true
shuffle: true
batchsize: 32
epochs: 50
dataset_config:
type: Heimdall.datasets.SingleInstanceDataset
head_config:
type: Heimdall.models.LinearCellPredHead
args: null
loss_config:
type: Heimdall.losses.FlattenCrossEntropyLoss
cell_rep_config:
type: Heimdall.cell_representations.CellRepresentation
scheduler:
name: cosine
lr_schedule_type: cosine
warmup_ratio: 0.1
num_epochs: 20
trainer:
type: Heimdall.trainer.HeimdallTrainer
args:
random_seed: 55
accumulate_grad_batches: 1
grad_norm_clip: 1.0
fastdev: false
skip_umaps: false
cpu: false
optimizer:
name: AdamW
args:
lr: 0.002
weight_decay: 0.1
betas:
- 0.9
- 0.95
foreach: false
fc:
type: Heimdall.fc.Fc
args:
max_input_length: 2048
embedding_parameters:
type: torch.nn.Module
tailor_config:
type: Heimdall.tailor.ReorderTailor
order_config:
type: Heimdall.order.ExpressionOrder
reduce_config:
type: Heimdall.reduce.SumReduce
fe:
type: Heimdall.fe.IdentityFe
name: Heimdall.fe.IdentityFe
args:
embedding_parameters:
type: Heimdall.embedding.ZeroBroadcast
args:
out_features: 128
d_embedding: 128
drop_zeros: true
fg:
type: Heimdall.fg.IdentityFg
args:
embedding_parameters:
type: Heimdall.embedding.FlexibleTypeEmbedding
args:
num_embeddings: vocab_size
embedding_dim: 128
d_embedding: 128
frozen: false
loss:
type: Heimdall.losses.FlattenCrossEntropyLoss
Training the model#
from Heimdall.trainer import setup_trainer
def training_loop(config):
trainer = setup_trainer(config, cpu=config.trainer.cpu)
if trainer is not None:
trainer.fit()
from accelerate import notebook_launcher
args = (config,)
notebook_launcher(training_loop, args, num_processes=1)